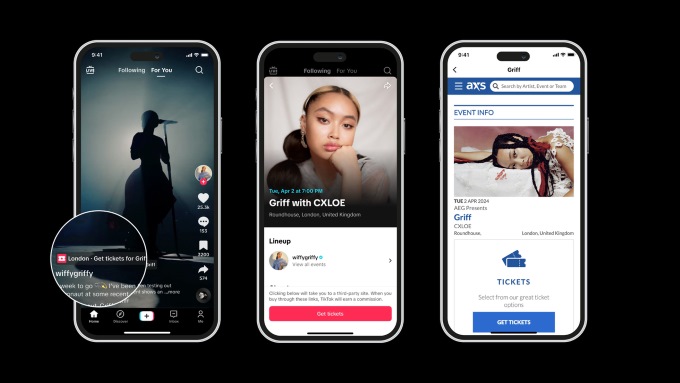
Beginning in May, select Apple stores around the country will offer a special Today at Apple series titled “Made for Business” that will give small business owners and entrepreneurs free opportunities to learn how Apple products and services can support their growth and success. Led by small business owners, the sessions will highlight how these […]